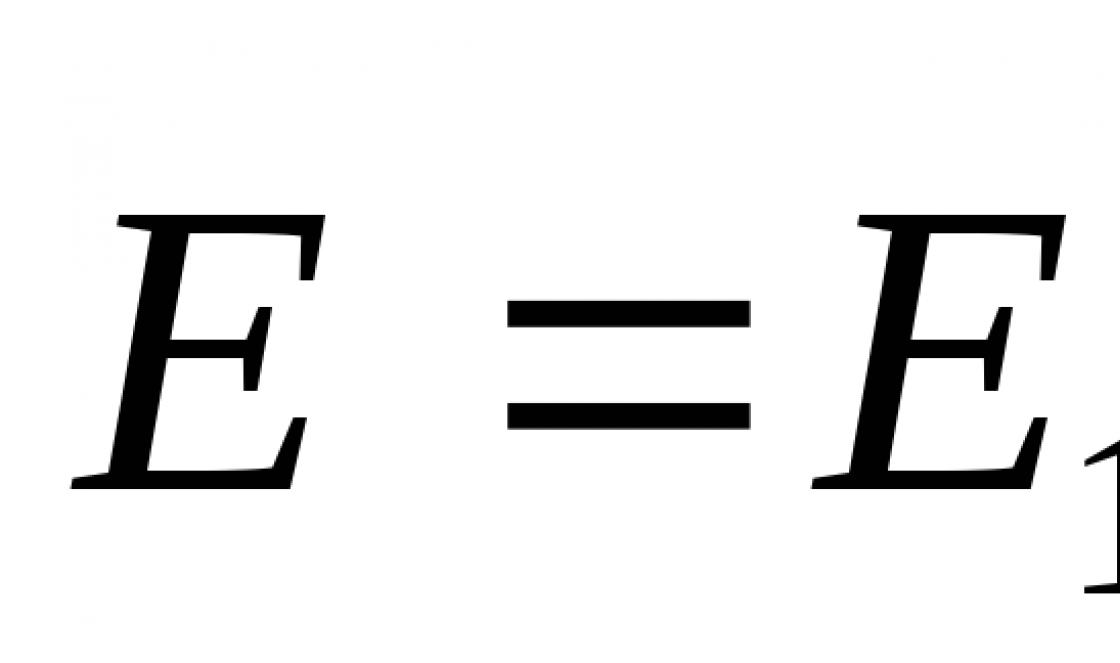Két atom közötti kölcsönhatás esetén:
U – kölcsönhatási energia;
U = U ELŐZETES. + U VISSZA
 - Lennard-Jones egyenlet
, c, b, m = állandó
- Lennard-Jones egyenlet
, c, b, m = állandó
Az atomok szilárd felülettel való kölcsönhatása esetén az összes kölcsönhatást összegezni kell.
x – felülettől való távolság
r – vonzóerők hatássugara
dV – hangerő
n – felületi molekulák száma
U HIRDETÉSEK. – az adszorpciós kölcsönhatás energiája



Adszorpció esetén a vonzás fokozódik. Nem poláris-nem poláris kölcsönhatás esetén pedig az adszorpció túlnyomórészt mélyedésekben lokalizálódik.
Elektrosztatikus kölcsönhatás.
Poláris adszorbens – nem poláris adszorbens
Nem poláris adszorbens – poláris adszorbens
Poláris adszorbens – poláris adszorbens.
M  Az adszorbens molekulát dipólusként, az adszorbenst pedig vezetőként ábrázoljuk, amelyben az adszorbátum molekula az adotthoz képest szimmetrikusan dipólustükröt indukál.
Az adszorbens molekulát dipólusként, az adszorbenst pedig vezetőként ábrázoljuk, amelyben az adszorbátum molekula az adotthoz képest szimmetrikusan dipólustükröt indukál.
X – távolság a közepétől
Az interakció során potenciál merül fel:
 ,
,
 - dipólusmomentum.
- dipólusmomentum.
A potenciál hajlamos felvenni a maximális értéket, pl. a dipólusok hajlamosak a felületre merőlegesen tájékozódni.
Mivel a hőmérséklet emelkedése elősegíti a Brown-mozgás növekedését, ez az adszorpciós folyamat gátlásához vezet.
Elektrosztatikus kölcsönhatás esetén az adszorbátum túlnyomórészt a kiemelkedéseken lokalizálódik.
Alapvető adszorpciós egyenlet.
Adszorpció esetén a komponens újraeloszlása következik be, ami azt jelenti, hogy a kémiai potenciál megváltozik. Az adszorpciós folyamat a felületi energia átalakulásának tekinthető kémiai energiává.
Rétegtérfogat = 0, akkor a termodinamika I. és II. törvényének általánosított egyenlete:
T = const; (1) = (2) => 

Kétkomponensű rendszer esetén:
,
 ,
,
 =>
=>
=>
 - Gibbs adszorpciós egyenlet
.
- Gibbs adszorpciós egyenlet
.
TV-adszorpció esetére. karosszéria - gáz: , 
 ,
,

 - izoterma
- izoterma
 - izobár
- izobár
 - izopiknális
- izopiknális
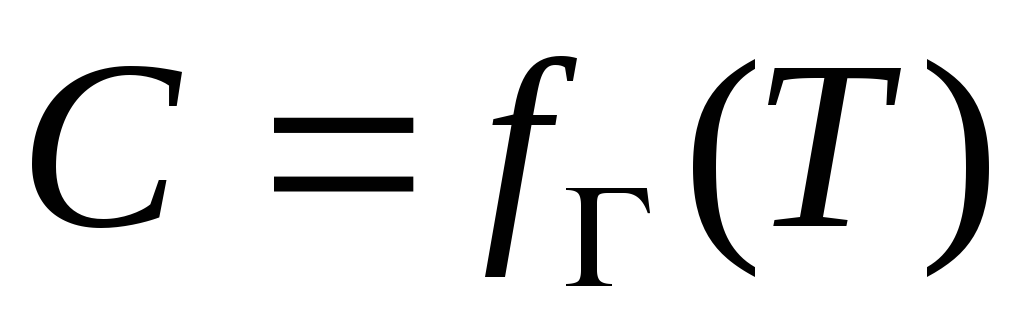 - izosztér
- izosztér
Az izoterma, az izopikn, az izosztér rokonságban állnak egymással.
Mert adszorpciós funkció 



Henry izoterma Langmuir izoterma



Termodinamika. Adszorpció.
Kondenzált anyagokhoz:
 ,
,
 ,
,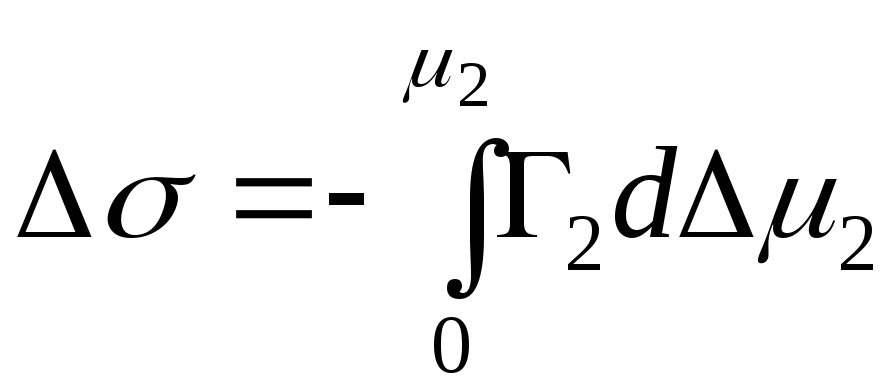
 - integrált változás a Gibbs-energiában
.
- integrált változás a Gibbs-energiában
.

P – ívelt felületre gyakorolt nyomás, Р S – sík felületre gyakorolt nyomás
 - adszorpciós potenciál
- adszorpciós potenciál
Differenciális változás a betáplálásban 
 , Г = állandó
, Г = állandó
- differenciális entrópia változás
- az adszorpció differenciális entalpiája
 - izoszterikus adszorpciós hő
- izoszterikus adszorpciós hő
 - kondenzációs hő
- kondenzációs hő
 - nettó adszorpciós hő
- nettó adszorpciós hő

 ,
,




Qa – integrált adszorpciós hő, 
Qra – integrált nettó adszorpciós hő,

Henry egyenlete
Az adszorpció vizsgálatát a felület heterogenitása nehezíti, így a homogén felületekre a legegyszerűbb törvényszerűségeket kapjuk.
Tekintsük a gázok szilárd felülettel való kölcsönhatását, amikor egy gáz térfogatában egyensúlyi állapotból a felületen egyensúlyi állapotba megy át. Ez az eset analóg a gázok egyensúlyi helyzetével a gravitációs térben.
 ,
,
 ,
=>
,
=> -Henry egyenlete
-Henry egyenlete
 - eloszlási együttható
- eloszlási együttható
Az adszorpciós folyamat során a kémiai potenciálok megváltoznak.
A tömeges fázishoz: 
A felszínen lévő gáz esetén: 
Egyensúlyi állapotban  , azaz
, azaz

Henry egyenletében az állandó nem függ a koncentrációtól
A Henry-egyenlet az alacsony nyomások és koncentrációk tartományában érvényes. A koncentráció növekedésével kétféle eltérés lehetséges a Henry-törvénytől:
1 – pozitív eltérések, D csökken, A csökken
2 – negatív eltérések, D – növekszik, A – növekszik.
Az eltérés típusát az adszorbens-adszorbátum kölcsönhatás egyik vagy másik típusának túlsúlya határozza meg.
Erős adhezív kölcsönhatás esetén az aktivitási együtthatók nőnek - ez pozitív eltérés. Kohéziós kölcsönhatások esetén negatív eltérések figyelhetők meg.
Monomolekuláris adszorpció.
Langmuir izoterma.
A legegyszerűbb mintákat Henry elméletében kaptuk. Langmuir egy olyan elméletet javasolt, amely szerint az adszorpciót kvázi-kémiai reakciónak tekintik. Ebben az esetben:
A felület energetikailag homogén.
Az adszorpció lokalizált, minden adszorpciós központ kölcsönhatásba lép egy adszorbált molekulával.
Az adszorbált molekulák nem lépnek kölcsönhatásba egymással.
Egyrétegű adszorpció.

 - felület,
- felület,  - adszorbeál,
- adszorbeál,  - adszorpciós komplex.
- adszorpciós komplex.
 , akkor az adszorpciós helyek koncentrációja:
, akkor az adszorpciós helyek koncentrációja:  ,
, - az adszorpció korlátozása.
- az adszorpció korlátozása.
 , akkor a reakcióállandó:
, akkor a reakcióállandó: 

 - Langmuir egyenlet.
- Langmuir egyenlet.
Az adszorpció függése a koncentrációtól
1 )
)
 ,
,
2) magas koncentrációjú terület
 - adszorpció korlátozása, monomolekuláris réteg kialakulása
- adszorpció korlátozása, monomolekuláris réteg kialakulása
Gibbs energiához: .
g az entrópiatényező.
A Henry-izoterma esetében a Gibbs-energia jellemzi az adszorbátum átmenetét az ömlesztett standard állapotból a felületen lévő standard állapotba. A Langmuir izoterma esetében  az adszorbens és az adszorbens közti affinitás mértékét jellemzi.
az adszorbens és az adszorbens közti affinitás mértékét jellemzi.
 találtak a van't Hoff isobarból.
találtak a van't Hoff isobarból.

 , Akkor
, Akkor  , innen
, innen  .
.

 - felületi töltési fok.
- felületi töltési fok.




 - szabad ülőhelyek száma,
- szabad ülőhelyek száma,  - elfoglalt helyek száma.
- elfoglalt helyek száma.
 ,
,

Azok. a nagy koncentrációk tartományában a szabad helyek száma fordítottan arányos az adszorbátum mennyiségével.
Gázelegy adszorpciója homogén felületen.
Ebben az esetben az adszorpciós folyamatot két párhuzamos reakciónak tekintjük.
 (1)
(1)

 (2)
(2)


Gázkeverék adszorpciója nem egyenletes felületen.
Egyenetlen felület esetén nem lehet az átlagos töltetekre korlátozódni.
A verseny eredményeként különböző típusú területeken különböző adszorbátumok lokalizálása lehetséges.
Ebben az esetben a kapcsolat  .
.
 ,
,
 - az adszorbátum telített gőznyomása.
- az adszorbátum telített gőznyomása.
 ,
,
 - adszorpciós hő.
- adszorpciós hő.
"+" - szimbátfüggőség, "-" - antibátfüggőség, "N" - nincs korreláció.
„+” - az adszorpció ugyanazzal a mechanizmussal megy végbe. Az energetikailag legkedvezőbb területeken túlnyomórészt a felülethez nagy affinitású gázok adszorbeálódnak.
„-” - az adszorpció különféle mechanizmusokon keresztül megy végbe, és egy bizonyos időpontig nincs verseny a felületért.
A monomolekuláris adszorpció túlnyomórészt a gázok kis értékű fizikai adszorpciója során valósul meg p, valamint a folyadék/gáz határfelületen.
Polimolekuláris adszorpció.
BET elmélet(Brunauer, Emmett, Teller).
Abban az esetben, ha az egyrétegű réteg képződése nem elegendő a felületi energia kompenzálásához, az adszorpció polimolekuláris, és a felületi erők hatására létrejövő kényszerkondenzáció eredményének tekinthető.
Főbb pontok:
Amikor egy adszorbált molekula eléri a foglalt helyet, többszörös halmaz jön létre.
Ahogy közelebb érünk p To p s csökken a szabad adszorpciós helyek száma. Kezdetben nő, majd csökken az egyesek, párosok stb. által elfoglalt helyek száma. készletekben.
at p =p s az adszorpció kondenzációvá alakul.
Nincsenek horizontális kölcsönhatások.
Az első rétegnél a Langmuir izoterma teljesül.
A felületet adszorpciós helyek halmazának tekintik. A dinamikus egyensúly feltétele érvényes: a szabad helyeken a kondenzáció sebessége megegyezik a foglalt helyekről történő párolgás sebességével.
a a kondenzációs együttható (a felületen kondenzált molekulák hányada);
 ,
,
Zm – a szabad helyek maximális száma.
 - az atomi rezgések frekvenciája a felületre merőleges irányban.
- az atomi rezgések frekvenciája a felületre merőleges irányban.
Az első réteghez dinamikus egyensúlyi feltételek:

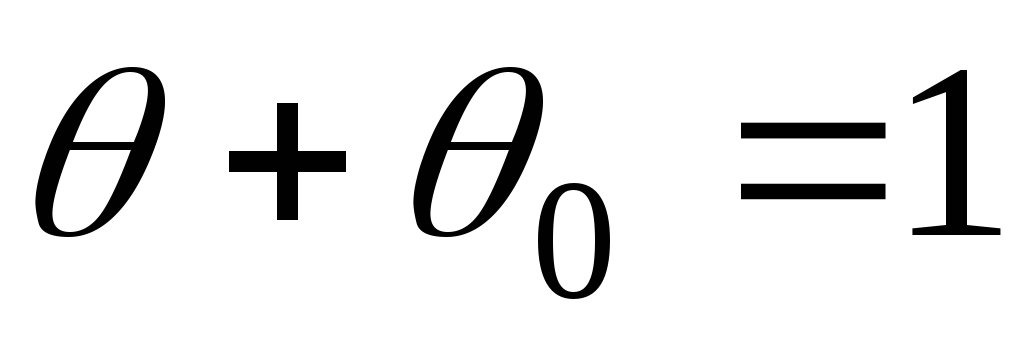

 , Akkor
, Akkor
 - Langmuir egyenlet.
- Langmuir egyenlet.
A második rétegre igaz lesz: 
Az i-edik réteghez: 
Az egyszerűség kedvéért feltételezzük, hogy a és ν minden rétegre azonos, kivéve az elsőt. Az első kivételével minden réteg esetében az adszorpciós hő állandó. Az utolsó rétegnél az adszorpciós hő megegyezik a kondenzációs hővel. Ennek eredményeként megkaptuk az egyenletet
 (*)
(*)
C- állandó,
A BET elmélet esetében az állandó VEL a tiszta adszorpció Gibbs-energiáját jellemzi. Az egyenlet csak egy állandót tartalmaz, és ez az egyenlet nagyon fontos az adszorbens fajlagos felületének meghatározásához.
Mivel az adszorpció következtében hő szabadul fel, alacsony hőmérsékleten határozzák meg a fajlagos felületeket.
 ????????????
????????????


Az elmélet fő hátránya– a horizontális kölcsönhatások figyelmen kívül hagyása a függőlegesek javára.
Az egyenlet a tartományban marad  0,05-től 0,3-ig.
0,05-től 0,3-ig.
Ahol  <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 > 0,3 – az adszorbátum – adszorbátum kölcsönhatás érintett.
> 0,3 – az adszorbátum – adszorbátum kölcsönhatás érintett.
Adszorbátum-adszorbátum kölcsönhatások számítása.
Kölcsönhatások akkor lépnek fel, amikor elágazó molekulák vagy molekulák adszorbeálódnak egy nem poláris felületen. Képes partnerek kialakítására. Ebben az esetben az adszorpciós izotermák alakja megváltozik.
A  az adszorbens nem poláris.
az adszorbens nem poláris.
Az 1. grafikon a gyenge adszorbátum-adszorbens kölcsönhatásoknak és az erős adszorbát-adszorbens kölcsönhatásoknak felel meg.
A 2. grafikon az erős adszorbátum-adszorbens és az erős adszorbát-adszorbens kölcsönhatásoknak felel meg.
A 3. grafikon az erős adszorbátum-adszorbens kölcsönhatásnak és a gyenge adszorbát-adszorbens kölcsönhatásnak felel meg.
 ,
,

Adszorbátummolekulák közötti kölcsönhatás esetén figyelembe kell venni az aktivitási együtthatók változásait. És ez az egyenlet így van felírva:
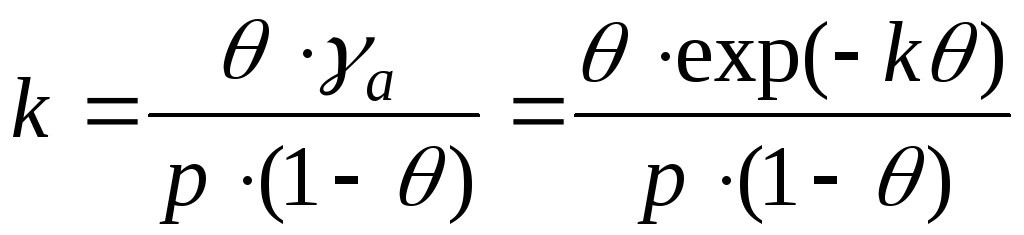 - Frunkin, Fowler, Guggenheim egyenlet.
- Frunkin, Fowler, Guggenheim egyenlet.
k– vonzási állandó.
Polyany potenciál elmélete.
Ez az elmélet nem vezet le semmilyen típusú adszorpciós izotermát, de lehetővé teszi az izotermák kiszámítását eltérő hőmérsékleten.
Adszorpció- ez az adszorpciós potenciál hatására az adszorbens felületéhez való vonzódásának az eredménye, amely nem függ más molekulák jelenlététől, és függ a felület és az adszorbens molekula távolságától.
 ,
,
 - adszorpciós potenciál.
- adszorpciós potenciál.
Mivel a felület nem egyenletes, a távolságot az adszorpciós térfogat helyettesíti  .Adszorpciós térfogat a felület és az adott értéknek megfelelő pont közé zárt térfogat
.Adszorpciós térfogat a felület és az adott értéknek megfelelő pont közé zárt térfogat  .
.
Adszorpciós potenciál az a munka, amely során 1 mól adszorbátumot egy adott adszorpciós térfogaton kívül az adszorpciós térfogat adott pontjára viszünk át (vagy 1 mól telített gőzt egy folyékony adszorbátummal egyensúlyban lévő adszorbens hiányában továbbítunk az adszorbenssel egyensúlyban lévő gőzfázisba).

Jellegzetes görbe
 - adszorpciós potenciál,
- adszorpciós potenciál, 
Egy adott adszorbensre és különböző adszorbátumokra a következő igaz: 
Különböző típusú adszorbátumokhoz  ,
,
Ahol  az adszorpciós izotermák potenciálja relatív nyomáson
az adszorpciós izotermák potenciálja relatív nyomáson  az 1. adszorbátum és a 2. adszorbátum esetében. Ez az arány állandó érték.
az 1. adszorbátum és a 2. adszorbátum esetében. Ez az arány állandó érték.
 - affinitási együttható
- affinitási együttható
A kapilláris kondenzáció elmélete.
Az adszorpciós folyamat lefolyása nagymértékben függ a porózus test szerkezetétől.
|
Mikroporózus | |
|
Átmeneti porózus | |
|
Makropórusos |
Mikroporózus szorbensek esetén az adszorpciós erők mezői átfedik egymást. A makropórusos szorbensek esetében a pórusok szállítócsatornaként működnek. A kondenzációs folyamatok az átmenetileg porózus testekben a legjelentősebbek. A kapilláris kondenzáció bizonyos értékeknél megindul pÉs  , amikor a felületi energia egy része már kompenzálva van. Szükséges feltétel, hogy a felület legyen önnedvesítő. A folyamat le van írva Thompson–Kelvin egyenlet.
, amikor a felületi energia egy része már kompenzálva van. Szükséges feltétel, hogy a felület legyen önnedvesítő. A folyamat le van írva Thompson–Kelvin egyenlet.

 - nedvesítés esetén a görbületi középpont a gázfázisban van.
- nedvesítés esetén a görbületi középpont a gázfázisban van.
Kapilláris kondenzáció esetén az adszorpciós izotermának hiszteretikus formája van. Az alsó ág az adszorpciós folyamatnak, a felső ág a deszorpciós folyamatnak felel meg.
Minden típusú pórus három típusra csökkenthető:
|
Kúpos |
Henger alakú, egy zárt véggel |
Henger alakú, két nyitott véggel |
|
A folyamatfeltöltés a pórusok aljáról történik. Az adszorpciós izoterma és a deszorpciós izoterma ebben az esetben egybeesik, mivel az adszorpciós folyamat egy gömbből indul ki, és a deszorpciós folyamat is néhány gömb eltűnésével kezdődik.
↓ |
Nincs hiszterézis. Az előre és hátra löketet a következő egyenlet írja le:
|
Alja nincs sehol, a pórusok kitöltése a henger falán megy majd.
henger: Az izoterma hisztérikus megjelenésű lesz.
↓ |


 - gömb,
- gömb, ,
,



IN  Nedvesedési körülmények között kisebb nyomáson kondenzáció lép fel, ami energetikailag kedvező. A deszorpciós ágból pórusméret-eloszlási görbéket kapunk.
Nedvesedési körülmények között kisebb nyomáson kondenzáció lép fel, ami energetikailag kedvező. A deszorpciós ágból pórusméret-eloszlási görbéket kapunk.
A differenciálgörbe maximumát az integrálgörbe inflexiós pontjához képest balra toljuk. A kis pórusok teljes térfogata kicsi, de nagy felülettel rendelkezik. A pórusméret növekedésével térfogatuk nő, mint  , és a környék olyan
, és a környék olyan  , ennek köszönhetően a differenciálgörbe maximumának eltolódása figyelhető meg.
, ennek köszönhetően a differenciálgörbe maximumának eltolódása figyelhető meg.
Adszorpció a szilárd-folyadék határfelületen.
A szilárd-gáz határfelületen történő adszorpció esetén egy komponenst figyelmen kívül hagytunk. A szilárd-folyadék határfelületen történő adszorpció esetén az adszorbátum kiszorítja az oldószermolekulákat az adszorbens felületéről.
 ,
,
Az egyenlet helyes:

 ,
,
N 1, N 2 – az oldószer és a komponens mólfrakciói, N 1 + N 2 = 1, majd
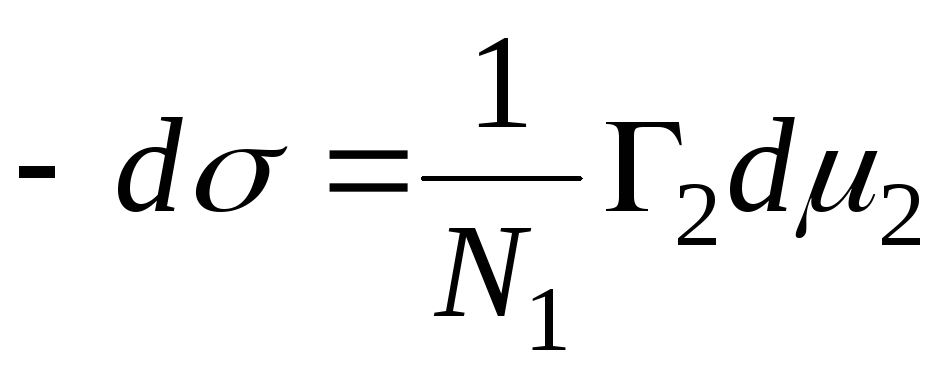 ,
=>
,
=>
 , akkor a szilárd-folyadék határfelület adszorpciós egyenlete.
, akkor a szilárd-folyadék határfelület adszorpciós egyenlete.
Adszorpció (G) > 0 at  <
0
<
0
Ha az értékek  mert a komponens és az oldószer nagyon eltérő, ebben az esetben a függőség G-tól Nértékén szélsőség van N
~ 0,5.
mert a komponens és az oldószer nagyon eltérő, ebben az esetben a függőség G-tól Nértékén szélsőség van N
~ 0,5.
E  ha
ha  közeli értékekkel rendelkeznek, ebben az esetben az adszorpció előjele változhat. Függőség G-tól N keresztezi az x tengelyt
közeli értékekkel rendelkeznek, ebben az esetben az adszorpció előjele változhat. Függőség G-tól N keresztezi az x tengelyt
Funkció metszéspontja G(N) az x tengellyel ún adszorpciós azeotróp. Ez azt jelenti, hogy a két komponens nem választható el egy adott adszorbensen.
Az adszorpciós izoterma egyenlete csereállandóval.
A szilárd-folyadék határfelületen történő adszorpció során a komponensek állandó újraeloszlása megy végbe az adszorbens felülete és az oldat térfogata között.
 - összetevők (- - felületre vonatkoznak)
- összetevők (- - felületre vonatkoznak)

 ,
,
 ,
, .
.
 ,
,


Adszorpció a folyadék-gáz határfelületen
R  Tekintsük a koncentrációprofil változását a folyadék-gáz határfelület átlépésével. Legyen a 2. komponens illékony.
Tekintsük a koncentrációprofil változását a folyadék-gáz határfelület átlépésével. Legyen a 2. komponens illékony.
Cs – koncentráció a felszíni rétegben.
A túlzott adszorpció definíciója alapján

Ha a komponens nem illékony, akkor az adszorpciós értéket a következőképpen írjuk fel:
P  ri
ri 

Az Eq.  egy anyag természetét a származéka írja le
egy anyag természetét a származéka írja le  .
.
A felületi feszültség izotermája 1-es vagy 2-es lehet:
1 – felületaktív anyagok
2 – felületaktív anyagok
A g felületi aktivitás az anyagok azon képessége, hogy csökkentsék a felületi feszültséget egy rendszerben.

 - a felületi réteg vastagsága
- a felületi réteg vastagsága
C s– a komponens koncentrációja a felületi rétegben
VEL– térfogatkoncentráció
A homológ sorozatokra van egy szabály:
 - Traubo Duclos szabály
- Traubo Duclos szabály
Egy homológ sorozat esetén az adszorpciós izoterma így néz ki:
A helyett G-t írunk, mivel a felületi rétegben túlzott az adszorpció.
Felületi feszültség izoterma:

 - tiszta oldószer felületi feszültsége.
- tiszta oldószer felületi feszültsége.
 - alapvető adszorpciós egyenlet;
- alapvető adszorpciós egyenlet;
 - Langmuir egyenlet.
- Langmuir egyenlet.
Oldjuk meg őket együtt:

- Shishkovsky egyenlet.
B– állandó a homológ sorozatra.
A- az egyik homológról a másikra való áttérés 3-3,5-szeresére nő 
![]()
1 – alacsony koncentrációjú terület
![]()
2 – átlagos koncentráció
3 – monomolekuláris réteg
A felületaktív anyagok difil molekulák, azaz. egy poláris csoportot és egy nem poláros szénhidrogéncsoportot tartalmaznak.
o a molekula poláris része.
| - a molekula nem poláris része.
Poláros oldószerben a felületaktív anyagok molekulái úgy vannak orientálva, hogy a molekula poláris része az oldószer felé nézzen, a nem poláris része pedig a gázfázisba kerül.
Shishkovsky egyenletében  , a homológ sorozatnál állandó.
, a homológ sorozatnál állandó.
A felületaktív hatás kezd megjelenni n>5. A monomolekuláris réteg koncentrációjánál magasabb koncentrációknál a felületaktív oldatokban micellizáció megy végbe.
Micella– amfifil felületaktív anyag molekulák aggregátumának nevezzük, melynek szénhidrogén gyökei magot alkotnak, a poláris csoportok pedig vizes fázisba alakulnak.
Micella massza – micellás massza.
H  molekulák száma – aggregációs szám.
molekulák száma – aggregációs szám.
Gömb alakú micellák
Micellizáció esetén egyensúly jön létre az oldatban
CMC – a micellaképződés kritikus koncentrációja.
Mivel a micellát külön fázisnak tekintjük:
Egy homológ sorozatra van egy empirikus egyenlet:
a– a funkciós csoport feloldódási energiája.
b – az adszorpciós potenciál növelése, adszorpciós munka metilén egységenként.
– az adszorpciós potenciál növelése, adszorpciós munka metilén egységenként.
A szénhidrogénmag jelenléte a micellákban lehetőséget teremt arra, hogy a vízben nem oldódó vegyületek feloldódjanak a felületaktív anyagok vizes oldatában, ezt a jelenséget szolubilizációnak nevezzük (ami oldódik, az a szolubilizálószer, a felületaktív anyag az oldódást elősegítő anyag).
Az iszap lehet teljesen nem poláris, tartalmazhat poláris és nem poláris részeket is, és felületaktív anyag molekulaként orientált.
Mindenesetre a szolubilizáció során a micelláris tömeg és az aggregációs szám növekedése nem csak a szolubilizátum felvétele miatt következik be, hanem az egyensúlyi állapot fenntartásához szükséges felületaktív molekulák számának növekedése miatt is.
A szolubilizálás annál hatékonyabb, minél kisebb a szolubilizátum molekulatömege.

 ~ 72 mN\m.
~ 72 mN\m.
 ~ 33 mN\m.
~ 33 mN\m.
A felületaktív anyagok hatékonysága a CMC értékétől függ.
2D felületi rétegnyomás

→ -felületfeszültségi erők.
- kétdimenziós nyomás.
A felületi réteg egy felületaktív oldat és egy tiszta oldószer felületi feszültségének különbségével egyenlő, tiszta felületre ható erő.
Az oldat és a felületi réteg között egyensúly jön létre



at  van egy terület, ahol
van egy terület, ahol  lineárisan függ a koncentrációtól.
lineárisan függ a koncentrációtól.
G [mol/m2].
 - egy mól anyag által elfoglalt terület
- egy mól anyag által elfoglalt terület
Ekkor a kétdimenziós nyomási izotermának lesz a formája 
 - kétdimenziós nyomási izoterma.
- kétdimenziós nyomási izoterma.
Függőség  S M-től:
S M-től:

at  - a kétdimenziós nyomás meredeken növekszik. at
- a kétdimenziós nyomás meredeken növekszik. at  kétdimenziós deformálódik, ami hirtelen növekedést okoz
kétdimenziós deformálódik, ami hirtelen növekedést okoz  .
.
A mindkét oldalon azonos fázisokkal határolt filmet kétoldalasnak nevezzük. Az ilyen filmekben az anyalúg állandó mozgása figyelhető meg.
Az 5 nm-nél kisebb vastagságú filmeket fekete filmeknek nevezzük.

Az adszorpciós rétegeknek két jellemzővel kell rendelkezniük: viszkozitás és könnyű mobilitás, folyékonyság és rugalmasság.
A Marangoni hatás öngyógyító.
Gibbs háromszög,  - túlnyomás.
- túlnyomás.
A film megnyúlt, és mivel a folyadék egy része elhagyta, a felületaktív anyagok a szabad térbe rohannak. Gibbs-háromszög.
A testek adszorpciós erejének hatása.
A film felületén mindig van egy adszorpciós réteg, amihez akkor
Langmuir egyenlet:


 kétdimenziós nyomásba
kétdimenziós nyomásba
 - a Shishkovsky-egyenlet analógja
- a Shishkovsky-egyenlet analógja
Elektrokinetikai jelenségek. Elektromos kétrétegű (EDL).
Gelemholtz modell. Gouy-Chapman elmélet.
1808-as repülés

U – alakú csövet, merítsen bele 2 elektródát. Megsértik a kommunikáló edények törvényét, és megváltozik a csőben lévő folyadék szintje - elektrokinetikus jelenségek.
Kinetikai jelenségek:
Elektroforézis
Elektroozmózis
Áramlási (áramlási) potenciál
Ülepedési potenciál
Az 1. és 2. ábra potenciálkülönbség alkalmazásakor keletkezik, a lyukasztás és a kolloid részecskék ülepedése potenciálkülönbség megjelenését okozza.
Elektroozmózis a diszperziós közeg mozgása egy álló, szórt fázishoz képest elektromos áram hatására.
Elektroforézis – ez a diszpergált fázisú részecskék mozgása egy álló diszperziós közeghez képest elektromos áram hatására.
P  Az elektrokinetikai jelenségek előfordulásának oka a töltések térbeli szétválása és a kettős elektromos réteg megjelenése.
Az elektrokinetikai jelenségek előfordulásának oka a töltések térbeli szétválása és a kettős elektromos réteg megjelenése.
Az elektromos kettős réteg lapos kondenzátor, az egyik lemezt potenciálmeghatározó ionok, a másikat ellenionok alkotják. Az ionok ugyanúgy szennyezettek, ahogy a potenciált meghatározó koionok az oldat térfogatába nyomulnak. A lemezek közötti távolság  . A potenciál lineárisan csökken, a potenciálkülönbség
. A potenciál lineárisan csökken, a potenciálkülönbség  .
.
A külső potenciálkülönbség nyírási modulus megjelenését okozza  a szilárd test felülete mentén ható, egységnyi területre eső erőpár.
a szilárd test felülete mentén ható, egységnyi területre eső erőpár.

Egyensúlyi állapotban a nyírási modulus megegyezik a viszkózus súrlódási modulussal (  ).
).

A mi körülményeink között  ,
,

 - Gelemholtz-Smalukowski egyenlet
- Gelemholtz-Smalukowski egyenlet
 - fáziseltolódás lineáris sebessége.
- fáziseltolódás lineáris sebessége.
E– elektromos térerősség.
 - potenciálkülönbség a lemezek között
- potenciálkülönbség a lemezek között
 - elektroforetikus mobilitás [m 2 /(V*s)].
- elektroforetikus mobilitás [m 2 /(V*s)].
A Helemholtz-modell nem veszi figyelembe a molekulák hőmozgását. A valóságban az ionok eloszlása a kettős rétegben bonyolultabb.
Gui és Chapman a DES következő okait azonosította:
Egy ion átmenete az egyik fázisból a másikba, amikor az egyensúly létrejön.
Szilárd fázisú anyagok ionizálása.
A felület kiegészítése a diszperziós közegben lévő ionokkal.
Polarizáció külső áramforrásból.
Az elektromos kettős réteg fuzzy vagy diffúz szerkezetű. Az ionok általában egyenletesen oszlanak el a diffúz rétegben.
A diffúz réteg elleninonokból áll, a réteg hosszát azok mozgási energiája határozza meg. Az abszolút nullához közelítő hőmérsékleten az ellenionok a lehető legközelebb állnak a potenciált meghatározó ionokhoz.
Danya elmélete két egyenletre épül:
Boltzmann egyenlet

 - az elektrosztatikus kölcsönhatás erői ellen dolgoznak.
- az elektrosztatikus kölcsönhatás erői ellen dolgoznak.
 - térfogati töltéssűrűség.
- térfogati töltéssűrűség.

Poisson-egyenlet 
Mivel az EDL vastagsága sokkal kisebb, mint a részecskeméret, és egy lapos EDL esetében a koordinátákhoz viszonyított derivált  És
És  megszűnik.
megszűnik. 
Az e y at y<<1 функцию можно разложить в ряд Маклорена:

Korlátozzuk magunkat a sorozat két tagjára, akkor:


 - A DEL vastagság az a távolság, amelynél a DEL potenciál csökken e egyszer.
- A DEL vastagság az a távolság, amelynél a DEL potenciál csökken e egyszer.
Minél alacsonyabb a hőmérséklet, annál kevesebb  . T→0-nál – lapos DEL. Minél nagyobb a koncentráció, minél több én, annál kevesebb
. T→0-nál – lapos DEL. Minél nagyobb a koncentráció, minél több én, annál kevesebb  .
.





 A „–” azt jelenti, hogy a potenciál a távolsággal csökken. =>
A „–” azt jelenti, hogy a potenciál a távolsággal csökken. =>
=>

 ,
,
 - a potenciál exponenciálisan csökken.
- a potenciál exponenciálisan csökken.
A felületi töltéssűrűség potenciálja: 
A felületi töltés ellentétes előjelű térfogati töltés, távolságra integrálva.




=>


Ahol a potenciál 2,7-szeresére csökken - 
Kétrétegű kapacitás 
Az elmélet hátránya, hogy nem veszik figyelembe a Helemholtz-réteg jelenlétét, i.e. nem veszi figyelembe  , innen erednek a hibák a fő paraméterek meghatározásában. Nem magyarázza meg a különböző természetű ionok hatását az elektromos kettős réteg vastagságára sem.
, innen erednek a hibák a fő paraméterek meghatározásában. Nem magyarázza meg a különböző természetű ionok hatását az elektromos kettős réteg vastagságára sem.
Stern elmélete. Kolloid micella szerkezete.
Az elektromos kettős réteg két részből áll: sűrű és diffúz. A potenciálképző ionok és a speciálisan adszorbeált ionok kölcsönhatása következtében sűrű réteg jön létre. Ezek az ionok általában részben vagy teljesen dehidratáltak, és a potenciált meghatározó ionokkal azonos vagy ellentétes töltéssel rendelkezhetnek. Ez az elektrosztatikus kölcsönhatási energia arányától függ  és fajlagos adszorpciós potenciál
és fajlagos adszorpciós potenciál  . A sűrű rétegű ionok rögzítettek. Az ionok másik része a diffúz rétegben található, ezek az ionok szabadok és mélyebbre tudnak mozogni az oldatban, azaz. nagyobb koncentrációjú területről alacsonyabb koncentrációjú területre. A teljes töltéssűrűség két részből áll.
. A sűrű rétegű ionok rögzítettek. Az ionok másik része a diffúz rétegben található, ezek az ionok szabadok és mélyebbre tudnak mozogni az oldatban, azaz. nagyobb koncentrációjú területről alacsonyabb koncentrációjú területre. A teljes töltéssűrűség két részből áll.

 - a Helmholtz réteg töltése
- a Helmholtz réteg töltése
 - Diffúz rétegtöltés
- Diffúz rétegtöltés
A felületen bizonyos számú adszorpciós központ van, amelyek mindegyike egy ellenionnal lép kölcsönhatásba. Egy ilyen kvázi-kémiai reakció állandója egyenlő:

 , Hol
, Hol  - az oldatban lévő ellenionok mólrésze
- az oldatban lévő ellenionok mólrésze
Helmholtz-eloszlás
A potenciál lineárisan csökken
Gouy potenciáleloszlás. Nincs sűrű réteg, a potenciál exponenciálisan csökken az értéktől 
Stern eloszlás.
Kezdetben a potenciálcsökkenés lineáris, majd exponenciális.
Amikor elektroforézis esetén elektromos mezőt alkalmazunk, akkor nem a szilárd fázis részecskéje mozog közvetlenül, hanem a szilárd fázis részecskéje, amely körülveszi az ionréteget. A DES megismétli a diszpergált fázisú részecske alakját. Potenciál alkalmazásakor a diffúz réteg egy része leszakad. A törésvonalat ún csúszó határ.
A diffúz réteg egy részének szétválása következtében a csúszási határon fellépő potenciált ún. elektrokinetikus potenciál(Zeta potenciál  ).
).
A diszpergált fázisú részecskéket ellenionréteggel és kettős elektromos réteggel körbevevő részecskéknek nevezzük micella.
A kolloid micellák írásának szabályai:


1-1 töltőelektrolit
T – diszpergált fázisú részecske.
Az AA a határ a sűrű és a diffúz részek között.
BB – csúszóhatár.
A csúszó határ egybeeshet az AA vonallal, de lehet, hogy nem.
Azt a pH-értéket nevezzük, amelynél a zéta potenciál nulla izoelektromos pont.
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. Többlet CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
(CaSO 4 m∙nCa 2+ 2( n - x)Cl - ) 2 x + x Cl - - micella jelölés.
CaSO 4 m – adalékanyag.
CaSO 4 m∙nCa 2+ – mag.
CaSO 4 m∙nCa 2+ 2( n - x)Cl - - részecske.
2. Na 2 SO 4 felesleg
Na 2 SO 4 ↔ 2 Na + + SO 4 2-
(CaSO 4 m∙nSO 4 2- 2(n-x)Na + ) 2x- 2xNa + - micella
CaSO 4 m – adalékanyag.
CaSO 4 m∙nSO 4 2 + – mag.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - részecske
Gelemholtz-Smoluchowski egyenlet

 - a határelmozdulás lineáris sebessége (elektrozmózisban).
- a határelmozdulás lineáris sebessége (elektrozmózisban).
 - potenciálkülönbség a kondenzátorlapokon (elektroozmózisban).
- potenciálkülönbség a kondenzátorlapokon (elektroozmózisban).


 - az oldat térfogati áramlási sebessége, S– a cella keresztmetszeti területe.
- az oldat térfogati áramlási sebessége, S– a cella keresztmetszeti területe.

E– elektromos térerősség.


 (elektrozmózishoz).
(elektrozmózishoz).
Az áramlási potenciálhoz:

 - potenciál
- potenciál
 - nyomás a membránon
- nyomás a membránon
Az elektroforetikus mobilitások és az elektroozmotikus mobilitások értékei általában kisebbek, mint a számítottak. Ez a következők miatt történik:
Relaxációs hatás (ha egy diszpergált fázisú részecske elmozdul, az ionos atmoszféra szimmetriája megbomlik).
Elektroforetikus gátlás (az ellenionok mozgásából adódó további súrlódás fellépése).
Áramvezetékek torzulása elektromosan vezető részecskék esetén.
A felületi feszültség és a potenciál kapcsolata. Lippmann egyenlet.
Az EDL képződése spontán módon megy végbe, a rendszer azon vágya miatt, hogy csökkentse felületi energiáját. Állandó körülmények között TÉs p A termodinamika első és második törvényének általánosított egyenlete így néz ki:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1. Lippmann egyenlet.
- 1. Lippmann egyenlet.
 - felületi töltéssűrűség.
- felületi töltéssűrűség.
 - differenciálkapacitás.
- differenciálkapacitás.
 - 2. Lippmann egyenlet.
- 2. Lippmann egyenlet.
VEL– kapacitás.
Oldjuk meg az 1. Lippmann-egyenletet és az alapvető adszorpciós egyenletet:
 ,
,


 , Akkor
, Akkor
 - Nernst egyenlet
- Nernst egyenlet
 ,
,
 ,
,




 - az elektrokapilláris görbe (ECC) egyenlete.
- az elektrokapilláris görbe (ECC) egyenlete.
IN  :
: , De
, De 
A kationos felületaktív anyagok (CPAS) csökkentik az EKC katódos ágát.
Az anionos felületaktív anyagok (APS) csökkentik az EKC anódos ágát.
A nemionos felületaktív anyagok (NSAS) csökkentik az ECC középső részét.
A szórt rendszerek stabilitása. Elszakadó nyomás.
A diszpergált rendszerek feloszthatók:
A termodinamikailag instabil rendszerek kinetikailag stabilak lehetnek a metastabil állapotba való átmenet miatt.
Kétféle stabilitás létezik:
Az ülepedési stabilitás (a gravitációhoz viszonyítva).
Aggregatív stabilitás. (tapadáshoz viszonyítva)
Alvadás a részecskék adhéziós folyamata, ami az aggregációs stabilitás elvesztéséhez vezet. A koagulációt a hőmérséklet, a pH, a keverés és az ultrahang okozhatja.
A koagulációt megkülönböztetik:
Megfordítható.
Visszafordíthatatlan.
A koaguláció elektrolitok bevezetésével történik.
A véralvadás szabályai:

Film- ez a rendszer két határfelület közötti része.
Elszakadó nyomás akkor fordul elő, ha a filmvastagság meredeken csökken a közeledő felületi rétegek kölcsönhatása következtében.

„-” - a filmvastagság csökkenésével a szétválasztó nyomás nő.
P 0 a nyomás az ömlesztett fázisban, amely a közbenső réteg folytatása.
P 1 – nyomás a filmben.
A stabilitás elmélete. DLFO (Deryagin, Landau, Fairway, Overbeck).
A DLFO elmélet szerint a szétválasztó nyomásnak két összetevője van:
Elektrosztatikus P E (pozitív, az elektrosztatikus taszító erők miatt van). Megfelel a Gibbs-energia csökkenésének a filmvastagság növekedésével.
Molekuláris P M (negatív, vonzóerők hatására).
Kémiai felületi erők hatására filmkompresszió okozza, az erők hatássugara tized nm körülbelül 400 kJ/mol energiával.:

 Teljes kölcsönhatási energia
Teljes kölcsönhatási energia
 - a rendszer aggregatívan stabil
- a rendszer aggregatívan stabil
P  - instabil rendszer
- instabil rendszer
pozitív komponens.
A növekedés a potenciális energia növekedésének köszönhető, amikor vékony filmeket összenyomnak. Nagy vastagságú filmeknél az ionenergia-többlet kompenzálódik, és megegyezik a diszperziós közeg térfogatában fellépő energiakölcsönhatásokkal.  (
( Ha
Ha  - film vastagság,
- film vastagság,


- ionsugár) a film elvékonyodása a benne lévő minimális felületi energiájú molekulák és ionok eltűnéséhez és redukciójához vezet. A szomszédos részecskék száma csökken, aminek következtében a filmben maradó részecskék potenciális energiája nő.

A DLVO elmélet a részecskék kölcsönhatását lemezek kölcsönhatásának tekinti.



 A részecskék nem lépnek kölcsönhatásba
A részecskék nem lépnek kölcsönhatásba  ,
,


- Laplace-egyenlet,
Gyengén töltött felületekhez

Erősen feltöltött felületekhez:
 ~
~
A molekuláris komponens két atom kölcsönhatása:

Egy atom kölcsönhatása felülettel:
Vegyünk két rekordot:  A molekuláris komponens megszerzéséhez össze kell összegezni a jobb és bal oldali lemezek atomjainak összes kölcsönhatási energiáját.
A molekuláris komponens megszerzéséhez össze kell összegezni a jobb és bal oldali lemezek atomjainak összes kölcsönhatási energiáját.
Ahol  - Hamaker állandó (figyelembe veszi a kölcsönható testek természetét).
- Hamaker állandó (figyelembe veszi a kölcsönható testek természetét).
Hogy. a részecskék kölcsönhatási energiája egy rendszerben potenciálgörbék segítségével fejezhető ki.
I – elsődleges potenciál minimum. Ez a visszafordíthatatlan koaguláció zónája, a vonzási erők érvényesülnek.
II – aggregatív stabilitás zónája, a taszító erők dominálnak.
III – szekunder potenciál minimum (vagy flokkulációs zóna). A diszpergált fázis részecskéi között elektrolitréteg van, a részecskék szétválaszthatók és az aggregációs stabilitási zónába vihetők át.

1. görbe – a rendszer aggregatívan stabil.
2. görbe – stabil az I. zónában, instabil a II.
3. görbe – koaguláció történt a rendszerben.
4. görbe – a 4. pontban a teljes kölcsönhatási energia U=0,  , ez az extrémum pont a gyors koaguláció kezdetének felel meg.
, ez az extrémum pont a gyors koaguláció kezdetének felel meg.
Két eset van:
1. Kissé feltöltött felületek:
U = U E + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - ez a koagulációs folyamat kezdetének megfelelő rétegvastagság.
- ez a koagulációs folyamat kezdetének megfelelő rétegvastagság.






 - gyengén töltött felületekhez
- gyengén töltött felületekhez
 Majd
Majd 
2. Erősen feltöltött felületekhez:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Nézzünk négyzetre (3)

Alvadás:
A specifikus adszorpció során az ionok szuperekvivalens mennyiségben adszorbeálhatók, így a felület megváltoztathatja a töltését. A felület feltöltődik.
Specifikus adszorpció esetén nemcsak ellentétes, hanem azonos előjelű ionok is adszorbeálódhatnak.
Ha a felülettel azonos előjelű ionok adszorbeálódnak, akkor a felületi rétegben nem csökken a potenciál, hanem nő.

Semlegesítő koaguláció (gyengén töltött részecskék részvételével történik, és nemcsak az elektrolit-koagulátor töltésétől, hanem a sűrű és diffúz rétegek határán lévő potenciáltól is függ).
Smoluchowski elmélete a gyors koagulációról.

A koagulációs sebesség függése az elektrolitkoncentrációtól.
I – alacsony a koagulációs ráta,
II – a véralvadási sebesség szinte arányos az elektrolitkoncentrációval.
III – gyors koaguláció régiója, a sebesség gyakorlatilag független a koncentrációtól.
Alapvető rendelkezések:
A kiindulási szol monodiszperz, a hasonló részecskék gömb alakúak.
Minden részecskeütközés hatékony.
Amikor két elsődleges részecske ütközik, egy másodlagos részecske képződik. Másodlagos + elsődleges = harmadlagos. Elsődleges, másodlagos, harmadlagos – multiplicitás.
Kémiai kinetika szempontjából a koagulációs folyamat a következő egyenlettel írható le:

A megoldás a következő egyenlet lesz: 
 - fele alvadási idő. Ez az az idő, amely alatt a szolrészecskék száma 2-szeresére csökken.
- fele alvadási idő. Ez az az idő, amely alatt a szolrészecskék száma 2-szeresére csökken.

 ,
,
 ,
,
 ,
,


A multiplicitás növekedésével a koagulációs görbék maximuma a nagyobb értékek felé tolódik el  .
.
Hibák:
A monodiszperzitás feltételezése.
Feltételezés az összes ütközés hatékonyságáról.
Adszorpciós folyamatok termodinamikája.
| Paraméter neve | Jelentése |
| Cikk témája: | Adszorpciós folyamatok termodinamikája. |
| Rubrika (tematikus kategória) | Oktatás |
Az adszorpciós folyamatok alapvető definíciói és osztályozási módszerei.
Az adszorpció olyan jelenségekre vonatkozik, amelyek a felületi energia spontán csökkenése miatt következnek be.
Adszorpció– egy heterogén rendszer összetevőinek spontán reverzibilis vagy irreverzibilis újraeloszlása a felületi réteg és a homogén fázis térfogata között.
A többkomponensű rendszerekben előnyösen a felületi feszültséget erősebben csökkentő komponens kerül át a felületi rétegre. Az egykomponensű rendszerekben a felületi réteg kialakulása során annak szerkezetében változás következik be (az atomok és molekulák bizonyos orientációja, polarizáció), ún. autoadszorpció.
A sűrűbb fázist, amelyen az adszorpciós kölcsönhatások lokalizálódnak, nevezzük adszorbens. A homogén fázis térfogata és a felületi réteg között újraeloszló anyagot a ʼʼ kifejezéssel jelöljük. adszorbeáljákʼʼ.
Egyes esetekben az adszorpciós folyamat visszafordítható. Ebben az esetben bizonyos körülmények között a molekuláris kinetikai jelenségek következtében adszorbeált molekulák egy része a felszíni rétegből a tömbfázisba kerülhet. Az adszorpció fordított folyamatát ún deszorpció.
Az adszorpciós folyamatok osztályozási módszerei.
Az adszorpciós folyamatok osztályozása a kölcsönhatásban lévő fázisok aggregációs állapota szerint. Figyelembe véve a szomszédos fázisok aggregált állapotától való függést, a következő típusú adszorpciós folyamatokat különböztetjük meg:
Gázok adszorpciója szilárd adszorbenseken;
Oldott anyagok adszorpciója a „szilárd-folyadék” és „folyadék-folyadék” határfelületeken;
Felületaktív anyagok adszorpciója a folyadék-gáz határfelületen.
Az adszorpciós folyamatok osztályozása az adszorbens és az adszorbátum közötti kölcsönhatás mechanizmusa szerint. Az adszorpció az adszorpátum molekulák és az adszorbens aktív központjai közötti kölcsönhatásnak tekinthető. Kölcsönhatásuk mechanizmusa szerint az adszorpció következő típusait osztják fel:
1) fizikai (molekuláris) adszorpció– az adszorbátum és az adszorbens molekulái közötti kölcsönhatás van der Waals erők, hidrogénkötések hatására jön létre (kémiai reakciók nélkül);
2) kémiai adszorpció (kemiszorpció)– az adszorbens molekulák az adszorbens aktív centrumaihoz való kapcsolódása különféle típusú kémiai reakciók eredményeképpen történik (az ioncsere reakciók kivételével);
3) ioncserélő adszorpció (ioncsere) – az adszorbált anyag újraeloszlása az oldat és a szilárd fázis (ioncserélő) között az ioncserélő reakciók mechanizmusa szerint.
Az adszorpciós folyamatok kvantitatív leírására két mennyiséget használunk.
1) Abszolút adszorpció– az adszorbens egységnyi felületére vagy tömegére jutó adszorbátum mennyisége (mol) vagy tömege (kg). Megnevezés – A; méret: mol/m2, mol/kg, kg/m2, kg/kᴦ.
2) Gibbs (túlzott) adszorpció– adszorbeált anyag feleslege egy bizonyos vastagságú felületi rétegben a homogén fázis térfogatában, egységnyi felületre vagy az adszorbens tömegére vonatkoztatva. Megnevezés – G; méret: mol/m 2, mol/kᴦ.
Az abszolút és a többlet adszorpció közötti összefüggés a következő egyenlettel szemléltethető:
Г = А – с * h (3,1)
ahol c az anyag egyensúlyi koncentrációja a fázis térfogatában, mol/m3;
h a felületi réteg vastagsága, hagyományosan 10-9 m-nek feltételezve.
Többkomponensű heterogén rendszerekben, amikor az egyik vagy másik komponens újraeloszlik a homogén fázis térfogata és a felületi réteg között, a felület többlet belső energiájának egyenlete érvényes:
U = T * S + s * s + Sm i * n i (3.2.)
Az egyenlet összes tagját a fázisközi felület egységnyi területére redukálva kapjuk:
U s = T * S s + s + Sm i * Г i (3.3)
ahol Г i = n i / s az i-edik komponens feleslege a felületi rétegben, vagyis a Gibbs-adszorpció.
Egykomponensű rendszer esetén a (3.3) egyenlet a következőképpen alakul:
G s = s + m * G (3,4)
ahol G s = U s - T * S s – a felület Gibbs-energiája vagy egységnyi felület létrehozásának munkája;
m * G – az adszorbeált anyag anyagának tömörödése a felületi rétegben.
A (3.4) egyenlet alapján arra a következtetésre juthatunk, hogy az adszorpció során a fázisközi felület létrehozásának munkája a felületképzés (az adszorbált fázis térfogatában lévő kohéziós kötések felszakítása) és az anyag felületi rétegben történő tömörítéséből áll.
Az adszorbens és az adszorbátum közötti dinamikus egyensúlyi állapotban a heterogén rendszer Gibbs-energiájának változása ΔG = 0, az adszorpciós folyamat termodinamikáját az ún. Gibbs alapvető adszorpciós egyenlet:
Ds = SГ i * dm i (3,5)
Ez az egyenlet univerzális, mivel minden típusú adszorpciós folyamatra érvényes
A Gibbs-adszorpciós egyenlet speciális esetei.
1) Adszorpció oldatokból.
A rendszer i-edik komponensének kémiai potenciáljára a „folyadék-szilárd adszorbens” és a „folyadék-gáz” határfelületeken történő adszorpció során a következő egyenletek érvényesek:
m i = m i 0 + R*T*ln a i (3,6)
dm i = R*T* d ln a i (3,7)
ahol m i 0 a rendszer i-edik komponensének kémiai potenciálja standard körülmények között;
a i a rendszer i-edik összetevőjének aktivitása normál körülmények között.
Ennek alapján a Gibbs-adszorpciós egyenlet a következőképpen alakul:
Г i = - a i / R*T * (ds / da i) (3.8)
Nem elektrolitok oldataihoz a i = c i-t vesszük, akkor:
Г i = - с / R*T * (ds / dс) (3,9)
Elektrolit oldatokhoz:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10.)
ahol с ± az oldat átlagos ionkoncentrációja;
n a sztöchiometrikus együttható.
2) Anyagok adszorpciója a gázfázisból.
A Mengyelejev-Clayperon egyenletnek megfelelően:
Р = с * R*T (3,11)
Ebben a tekintetben a szilárd adszorbenseken lévő gázok adszorpciójára vonatkozó Gibbs-egyenlet a következő formában van felírva:
Г i = - Р / R*T * (ds / dР) (3,12)
A gyakorlatban a Gibbs-adszorpciós egyenlet lehetővé teszi a folyadékkoncentráció vagy az egyensúlyi gáznyomás különböző értékeinél végzett felületi feszültség mérések alapján, hogy kiszámítsuk a felületi rétegben lévő anyagok adszorpciójának mennyiségét, amelyre a felületi feszültséget meghatározzák.
Adszorpciós folyamatok termodinamikája. - koncepció és típusok. Az "Adszorpciós folyamatok termodinamikája" kategória osztályozása és jellemzői. 2017, 2018.
Az adszorpció a felületen megy végbe. Ezért indokolt a felületi jelenségek termodinamikai leírását a heterogén rendszerek termodinamikájának speciális esetének tekinteni.
Rizs. 3.4. Gibbs adszorpció: 1 - kétfázisú összehasonlító rendszer, 2 - valódi kétfázisú rendszer, nem egyenletes régióval
A heterogén rendszerek termodinamikájában alkalmazzák additívitás elve ami a következő: egy heterogén rendszer minden extenzív tulajdonsága egyenlő azoknak a megfelelő extenzív tulajdonságoknak az összegével, amelyekkel a fázisok rendelkeztek volna az érintkezésük előtt. Jelöljük a fázisokat α-val és β-val (4. ábra). Ekkor egy ideális rendszerre, ahol a határfelülethez közeli fázisok tulajdonságai egybeesnek tömegtulajdonságaikkal, az alábbi összefüggések érvényesek az U belső energiára, V térfogatra, tömegre (mólszámra) n, S entrópiára, miután egyensúlyba lépett. heterogén rendszer:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
Ez azt feltételezi, hogy a hőmérséklet és a nyomás mindkét fázisban azonos.
Valódi heterogén rendszerek esetén a két fázis határán lévő átmeneti tartomány további hozzájárulást jelent a rendszer kiterjedt tulajdonságaihoz. Ha felszíni jelenségek fordulnak elő, akkor figyelembe kell venni a különbséget egy valódi heterogén rendszer extenzív tulajdonságai és egy olyan modellrendszer extenzív tulajdonságai között, amelyben a felületi jelenségek hiányoznak. Az ilyen rendszert összehasonlító rendszernek nevezzük. Az összehasonlító rendszer ugyanazokkal az intenzív paraméterekkel (T, P, C i ...) és V térfogattal rendelkezik, mint a valós rendszer (4. ábra).
Termodinamikai szempontból G adszorpciós érték alatt az n s anyag azon többletmennyiségét értjük, mólban vagy grammban kifejezve, amellyel egy valódi heterogén rendszer a referenciarendszerhez képest a határfelülethez vagy a felülethez viszonyítva rendelkezik. Feltételezzük, hogy az összehasonlító rendszer ugyanazokkal az intenzív paraméterekkel (T, P, C i) és térfogatával (V = V α + V β) rendelkezik, mint a valós rendszer (4. ábra) .
Г = (n - n α - n β)/A = n s /A 3.11
Valós rendszer átmeneti tartományának termodinamikai többletfüggvényei (ezeket s indexszel jelöljük) így írhatók fel.
U s = U - U α - U β , n s = n - n α - n β , S s = S - S α - S β stb.
Az adszorpció kísérleti mérései mindig pontosan úgy adják meg az adszorpciót, mint a komponens többletét a valós rendszerben a választott referenciarendszerhez képest. Például gáz adszorpciója szilárd adszorbensen vagy komponensek szilárd fázison történő adszorpciója esetén az adszorpciós értékek meghatározásához határozza meg az adszorbátum kezdeti koncentrációjának változását az α és β fázisok érintkezése után.
n i s = V(C i o - C i),
Ahol C i o– az i-edik komponens kezdeti koncentrációja, C i– az i-edik komponens koncentrációja az érintkező fázisok közötti egyensúly megteremtése után. Úgy tartják, hogy a kötet V nem változik. Azonban a koncentráció én komponens C i, amelyet kísérleti úton kapunk, térfogatban határozzuk meg V' fázis határfelülete felett anélkül, hogy figyelembe vennénk az átmeneti réteg inhomogén régiójának térfogatát V α azon a határfelületen, ahol a koncentráció van C i α. Így egy valós rendszerben egy nem egységes régió létezése miatt a rendszer teljes térfogata így ábrázolható. V = V’ + V α. Minden mennyiség én-edik komponens C i o e két kötet között lesz elosztva:
V C i o = V’ C i + V α C i α ,
és a komponens mólszáma én, a felületen adszorbeált, egyenlő lesz
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
Azok. a kísérletileg meghatározott adszorpció az i-edik komponens feleslege a V α térfogatban, összehasonlítva ennek a komponensnek a fázishatártól távol eső térfogatban lévő mennyiségével. Ezt a típusú adszorpciót Gibbs-adszorpciónak nevezik. .
V α C i α teljes tartalomnak nevezzük én- komponens az adszorpciós rétegben. Nagyon alacsony koncentrációk tartományában C i kötetben V' módosítás V α C i a (3.2) egyenlet figyelmen kívül hagyható és a mért érték figyelembe vehető V α C i α teljes tartalom én- komponens az adszorpciós rétegben, például szilárd adszorbensre történő gázadszorpció során alacsony nyomáson.
Kuznetsova E.S. és Buryak A.K. Összehasonlította az aminosavak és a társult adszorpció termodinamikai jellemzőit. A munka azt vizsgálta, hogy az aminosavak szerkezete, dimerjeik és az eluens komponensekkel asszociált vegyületei hogyan befolyásolják adszorpciójukat a szénanyagok felületén. A grafitizált termikus szén (GTS) felületén aromás aminosavak (fenilalanin, tirozin), heterociklusos aminosav (triptofán) és dimerjeik trifluor-ecetsavval (TFA) történő adszorpció (TCA) termodinamikai jellemzőinek molekuláris statisztikai számítását végeztük. ki. A kapott adatokat összehasonlítjuk a porózus grafitizált szén-hiperkarb aminosav-visszatartási mintázatával fordított fázisú, nagy teljesítményű folyadékkromatográfiás (RP HPLC) körülmények között. Kimutatták, hogy ezeknek a vegyületeknek a szénláncának növekedésével a TCA és az aminosav-visszatartási értékek nőnek.
Shkolin A.V. és Fomkin A.A. a metán-mikropórusos szénadszorbens AUK adszorpciós rendszer termodinamikai függvényeinek viselkedését (differenciális moláris izoszterikus adszorpciós hő, entrópia, entalpia és hőkapacitás) elemezték az adszorpciós egyensúlyi paraméterek függvényében a 65-7 °C közötti hőmérséklet-tartományban. 393 K és 1 Pa és 6 MPa közötti nyomások. A gázfázis nemideálisságának és az adszorbens nem tehetetlenségének befolyásának figyelembevétele az izoszterikus adszorpcióhő hőmérséklet-függésének megjelenéséhez vezetett, különösen az adszorbens nagy nyomásának tartományában. A vizsgált rendszer esetében az adszorpciós rendszer termodinamikai funkcióira a fő hatást a gázfázis nemideálissága gyakorolja. Az adszorbens nem tehetetlenségének korrekciója az adszorpciós rendszer paramétereinek ezen tartományában nem több, mint 2,5%.
Az Üzbég Köztársaság Tudományos Akadémia Általános és Szervetlen Kémiai Intézetében Muminov S.Z. munkájában a montmorillonit felületi tulajdonságainak és porózus szerkezetének változásait vizsgálta, amikor az ásvány cserélhető kationjait polihidroxi-alumínium kationokra cserélték. Az előzetes termikus vákuumozás jelentős hatással van a polihidroxialumínium-montmorillonit adszorpciós tulajdonságaira a metil-alkoholhoz viszonyítva. A dehidratált nátriumon és módosított montmorilloniton mért CH3 adszorpciós izoszterek sorozatából származó, széles hőmérsékleti tartományban mért adatok alapján megállapítottam az adszorpciós hő függését az adszorbeált anyag mennyiségétől.
N.S. Kazbanov, A.V. Matveeva és O.K. Krasilnikov tanulmányt végzett a fenol vizes oldatokból történő adszorpciójáról aktív szenek, például FAS, PAH és szénfilc által 293, 313 és 343 K hőmérsékleten, 5-250 mmol/l koncentrációtartományban. A szűk pórusméret-eloszlással jellemezhető szekvenciálisan aktív szén FAS minták sorozatát furfurol alapú polimerek karbonizálásával kaptuk. A PAH egy mikroporózus polimer aktív szén. A szénfilc hidratált cellulózszálak alapú rostos anyag. Az adszorbensek porózus szerkezetének paramétereit nitrogéngőz adszorpciós izotermákból határoztuk meg 77 K-en (ASAP-2020, Micromeritics, USA). Az oldatok adszorpciójának vizsgálatát ampulla módszerrel termosztátban végeztük. A kiválasztott mintákat spektrofotometriával elemeztük. A kapott folyadékfázisú adszorpciós izotermák elemzését a mikropórusok térfogati kitöltésének (VFM) elméletével végeztem a Dubinin-Radushkevich (DR) egyenlet szerint.
A hőmérséklet hatása a folyékony oldatok szorpciójára nem egyértelmű. Egyrészt a mikropórusos adszorbensek esetében a molekulák behatolása a méretükben ezekhez a molekulákhoz hasonló pórusokba a kinetikus energiától függ, és ennek megfelelően növekszik a hőmérséklettel. Másrészt a fizikai adszorpció exoterm folyamat, és az adszorpció a hőmérséklettel csökken. Ezen tényezők közötti kapcsolat minden rendszer esetében meghatározza az adszorpció hőmérséklet-függésének menetét.
Az adszorbens-fenol rendszer egyedisége abban rejlik, hogy az adszorpciós izotermák inverz hőmérséklet-függését mutatják. A hőmérséklet 293 K-ról 313 K-ra emelkedésével az adszorpció határértéke nő, ami nyilvánvalóan a molekulaszita hatásnak köszönhető: a hőmérséklet emelkedésével a fenolmolekulák a szénanyagok szűkebb pórusaiba is képesek behatolni. Az adszorpció főként mikropórusokban megy végbe, mivel az adszorbenseknek kis számú mezopórusa van. A mikropórusméret növekedésével a maximális adszorpciós értékek jelentősen megnövekednek, elérve a 2,9 mmol/g-ot PAH-nál, 8,5 mmol/g-t a FAS-nál és a 12,7 mmol/g-ot filcnél. A kapott adszorpciós izotermákat jól leírja a DR egyenlet, amelynek kitevője 2.